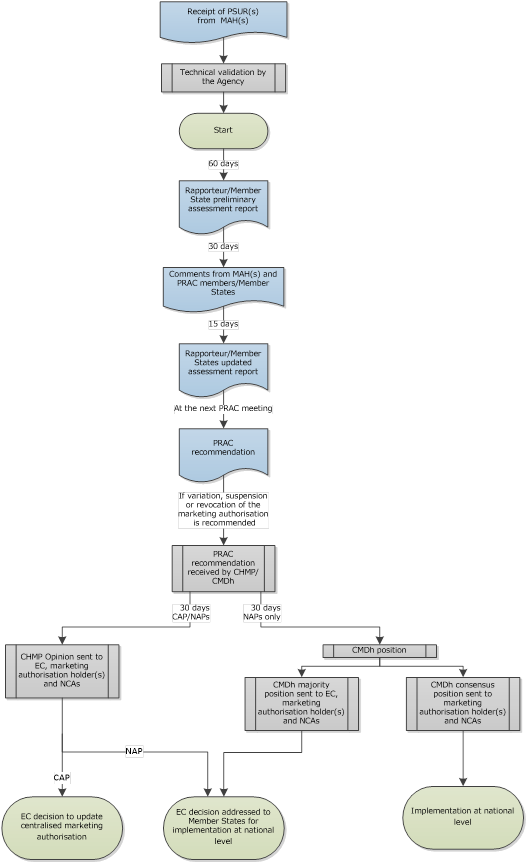
Periodic safety update reports (PSURs)
This page includes information on periodic safety update reports (PSURs), PSUR submission requirements, PSUR single assessment procedures (PSUSAs) and the European Union reference dates (EURD) list.
HumanRegulatory and procedural guidancePharmacovigilance