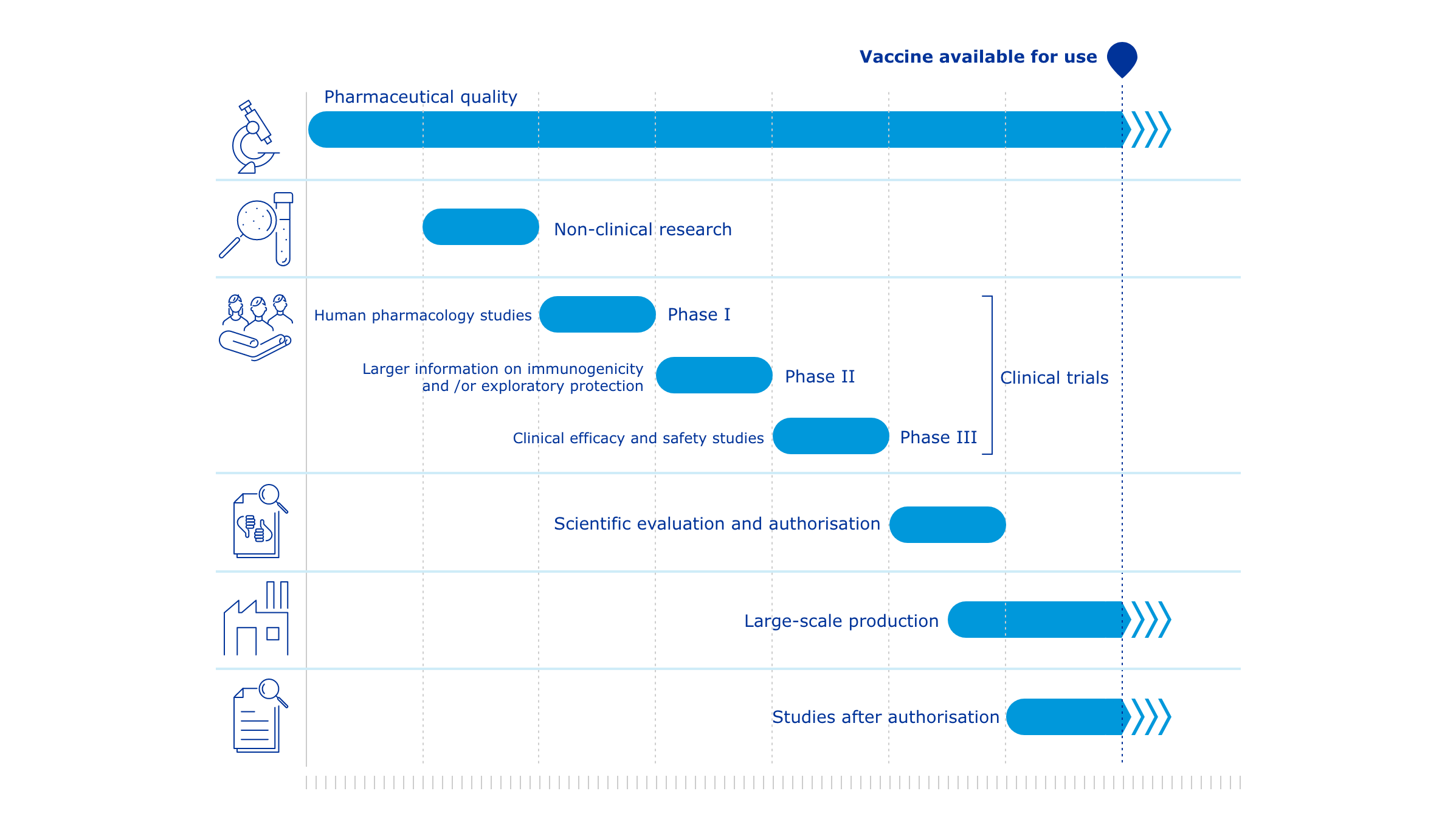
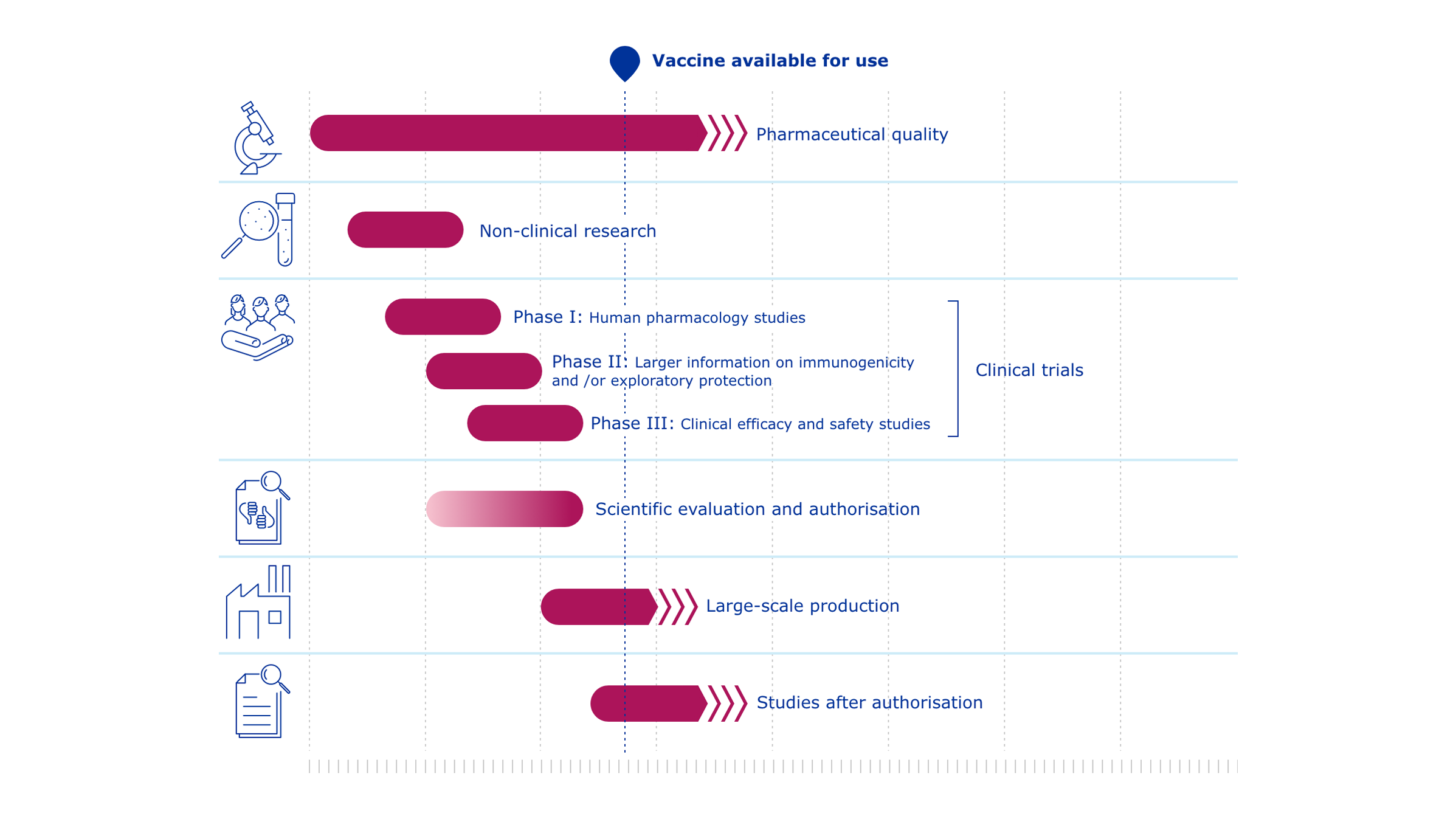
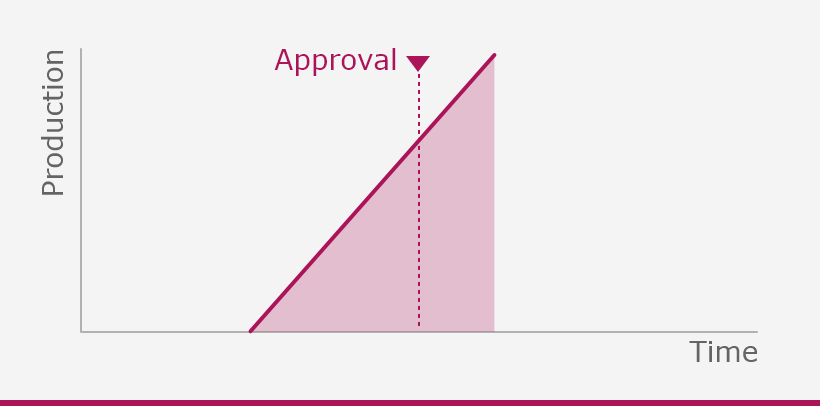
COVID-19 vaccines: development, evaluation, approval and monitoring
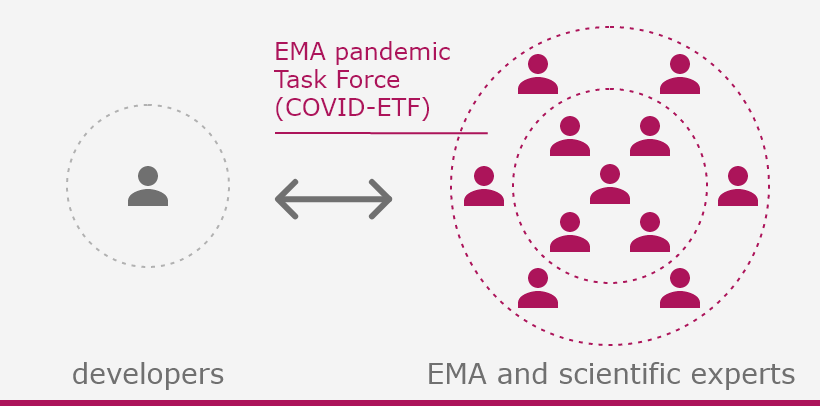
The European Medicines Agency (EMA) plays an important role in enabling the development, scientific evaluation, approval and monitoring of COVID-19 vaccines in the European Union (EU).
HumanCOVID-19Vaccines