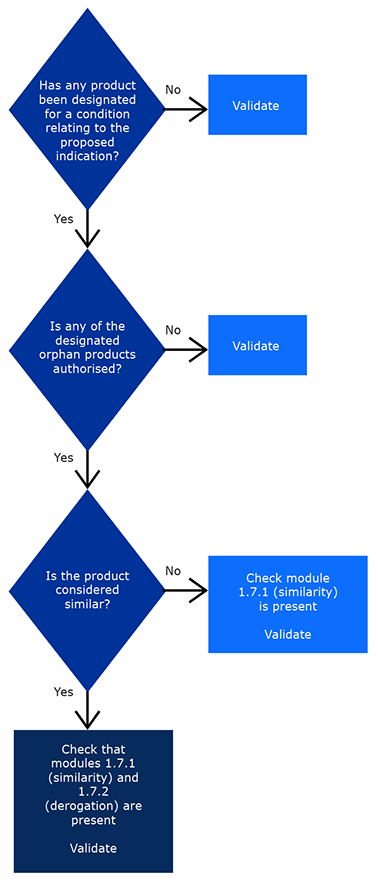
Pre-authorisation guidance
These questions and answers (Q&As) provide an overview of the European Medicines Agency's (EMA) advice on issues that are typically addressed in discussions or meetings with marketing authorisation holders in the application phase.
HumanRegulatory and procedural guidance